Abstract
Background: Human amniotic mesenchymal stem cells (hAMSCs) hold broad promise for cell therapy and regenerative medicine, yet replicative and stress-induced senescence during ex vivo expansion severely restricts their clinical application. We previously identified Schlafen family member 11 (SLFN11) as a gene that protects hAMSCs from DNA-damage–driven senescence, but its downstream effector target remained unknown.
Methods: hAMSCs were isolated by two-step enzymatic digestion and validated by immunocytochemistry and flow cytometry. A camptothecin (CPT)–induced DNA-damage senescence model was established. SLFN11 was stably overexpressed by lentiviral transduction, and its interactome was screened by immunoprecipitation coupled with mass spectrometry (IP–MS) and validated by co-immunoprecipitation (Co-IP). NONO was silenced by siRNA in SLFN11-overexpressing cells, and senescence, stemness, cell-cycle and proliferation markers were quantified. Three SLFN11 truncation mutants were generated to map the NONO-binding domain, complemented by reverse Co-IP and AlphaFold2/ZDOCK molecular docking.
Results: IP–MS identified 14 SLFN11-interacting proteins, with the RNA-binding protein NONO ranking highest by mass-spectrometry score. SLFN11 overexpression reduced SA-β-gal positivity, upregulated stemness factors (Nanog, Oct4, Sox2), suppressed p16/p21, and enhanced proliferation. Silencing NONO abolished each of these protective effects. All three SLFN11 domains bound NONO, but the C-terminal helicase domain (577–901 aa) displayed the strongest interaction, a finding corroborated by reverse Co-IP and docking.
Conclusions: NONO is the essential effector through which SLFN11 delays hAMSCs senescence, engaging predominantly via the SLFN11 C-terminal domain. The SLFN11–NONO axis represents a candidate node for mitigating stem-cell senescence and improving the efficacy of stem-cell–based therapies.
Keywords: SLFN11; human amniotic mesenchymal stem cells; cellular senescence; NONO; protein domain mapping
1Introduction
Ageing is a progressive, multifactorial decline in cellular and tissue function that ultimately compromises organismal viability. At the cellular level, senescence reflects the accumulation of diverse forms of damage that drive functional impairment and a deterioration in quality of life. Since its first description by Hayflick and Moorhead, cellular senescence has been increasingly recognised as a tractable therapeutic axis: selectively targeting senescent cells can ameliorate age-associated dysfunction in multiple tissues.
Stem-cell senescence and exhaustion are now regarded as primary hallmarks of ageing. The decline in the proliferative, differentiation, and regenerative capacity of adult stem cells is a major contributor to organismal ageing, and senescent stem cells exhibit attenuated self-renewal, restricted multilineage potential, and dysregulated autophagy and metabolism. Among adult stem cells, mesenchymal stem cells (MSCs) are particularly attractive owing to their wide tissue distribution and ease of isolation. Human amniotic mesenchymal stem cells (hAMSCs), derived from the amniotic membrane of full-term placenta, are especially valuable because they are abundant, easily obtained with maternal consent, raise minimal ethical concerns, and possess low immunogenicity. These properties have made hAMSCs a promising tool for cell therapy and regenerative medicine. However, hAMSCs progressively senesce during in vitroexpansion — manifested as reduced proliferation, an enhanced senescence-associated secretory phenotype (SASP), and diminished differentiation potential — which markedly limits their clinical deployment. Elucidating the intrinsic mechanisms of hAMSCs senescence is therefore of considerable importance.
In earlier work, our laboratory unexpectedly isolated a “non-senescent” hAMSC line (NS-hAMSCs) from placental tissue that retained stable proliferative capacity beyond passage 30 without typical senescence features. Genome-wide transcriptome profiling, with RT-PCR validation, nominated Schlafen family member 11 (SLFN11) as a key candidate gene underlying this senescence-resistant phenotype. Knockdown of SLFN11 accelerated senescence in NS-hAMSCs, whereas overexpression in ordinary hAMSCs delayed both long-term expansion- and camptothecin (CPT)–induced senescence, preserving proliferation and stemness, lowering cell-cycle–arrest proteins, and mitigating DNA damage. Nevertheless, the molecular target and mechanism through which SLFN11 delays hAMSCs senescence remained undefined.
A preliminary IP–MS screen following SLFN11 overexpression detected 14 interacting proteins, including NONO, DDX1, and G3BP2. The non-POU domain-containing octamer-binding protein (NONO) ranked highest by mass-spectrometry score and has established roles in transcriptional regulation, DNA-damage repair, proliferation, the immune response, and cell-cycle control — making it a compelling candidate effector. SLFN11 itself is a 901-residue human Schlafen family member that localises to the nucleus as a dimer and participates in proliferation, DNA replication, and the innate antiviral response. It comprises three principal domains: an N-terminal endonuclease domain (1–353 aa), a central linker domain (354–576 aa), and a C-terminal helicase domain (577–901 aa); the C-terminus specifically recognises the RPA1 subunit to dissociate RPA from single-stranded DNA, blocking checkpoint activation and homologous-recombination repair. The specific domain through which SLFN11 exerts its anti-senescence function in hAMSCs, however, was unknown.
Here, using hAMSCs as the model system, we screen and validate the SLFN11 interactome, demonstrate that NONO is the essential effector required for SLFN11-mediated delay of senescence, and map the SLFN11–NONO interaction to the SLFN11 C-terminal domain. These findings expand the regulatory framework of hAMSCs senescence and define the SLFN11–NONO axis as a potential target for anti-senescence intervention.
2Materials and Methods
2.1 Isolation and culture of hAMSCs
Amniotic membranes were obtained from healthy full-term caesarean deliveries after written informed consent, under approval of the Ethics Committee of the Affiliated Hospital of Zunyi Medical University (KLLY-2023-137). Donors were free of hepatitis B/C, HIV, and syphilis and were not of advanced maternal age. The amnion was mechanically peeled from the placenta under sterile conditions and digested using a two-step trypsin–collagenase protocol (0.5% trypsin–0.2% EDTA followed by 0.5 mg/mL type II collagenase with 0.05 mg/mL DNase I). Released cells were cultured in low-glucose DMEM/F12 supplemented with 10% NBCS, 10 ng/mL bFGF, and 1% penicillin–streptomycin at 37 °C in 5% CO2.
2.2 Phenotypic identification
Cell identity was confirmed by immunocytochemistry for the mesenchymal marker vimentin and the epithelial marker cytokeratin-19 (CK19), with PBS as a negative control. Surface immunophenotype was assessed by flow cytometry for the positive markers CD105, CD73, CD90, CD44, and CD29 and a haematopoietic/negative cocktail (CD34, CD11b, CD19, CD45, HLA-DR).
2.3 CPT-induced DNA-damage senescence model
Camptothecin (CPT), a topoisomerase-I inhibitor that generates double-strand breaks, was used to induce senescence. hAMSCs were treated with CPT (0.1–10 µM) for 2 h; the drug was then removed and cells were cultured for a further 24 h. The optimal concentration was determined by senescence-associated β-galactosidase (SA-β-gal) staining and CCK-8 viability assay, identifying 1 µM as the senescence-inducing dose with negligible cytotoxicity.
2.4 Lentiviral overexpression and Western blot
Passage-3 hAMSCs were transduced with lentivirus encoding EF1-EGFP-F2A-Puro-CMV-SLFN11-HA or the empty vector. The optimal multiplicity of infection (MOI) was determined 72 h post-infection by fluorescence intensity, and overexpression efficiency was verified by Western blot. Proteins were resolved by SDS-PAGE, transferred to PVDF membranes, probed with the indicated primary antibodies (SLFN11, NONO, Nanog, Oct4, Sox2, p16, p21, PCNA, GAPDH) and HRP-conjugated secondaries, and detected by ECL. Band intensities were normalised to GAPDH and quantified across three independent experiments.
2.5 IP–MS and co-immunoprecipitation
To identify SLFN11-interacting proteins, lysates from CPT-treated SLFN11-overexpressing hAMSCs were immunoprecipitated with an anti-SLFN11 antibody and analysed by liquid chromatography–tandem mass spectrometry (LC-MS/MS). Candidate interactions were validated by Co-IP: lysates were incubated with anti-SLFN11 (1:60) at 4 °C, captured on Protein A/G magnetic beads, and immunoblotted for candidate partners.
2.6 siRNA-mediated NONO knockdown
siRNA targeting NONO was transfected into hAMSCs; knockdown efficiency was confirmed by Western blot. In rescue experiments, SLFN11 was stably overexpressed and NONO was then silenced; following CPT-induced senescence, cells were assayed for SA-β-gal positivity, stemness factors (Nanog, Oct4, Sox2), cell-cycle proteins (p16, p21), and proliferation (PCNA Western blot and EdU incorporation).
2.7 Domain mapping and molecular docking
Three Flag-tagged SLFN11 truncations — N-terminal (1–353 aa), linker (354–576 aa), and C-terminal (577–901 aa) — and an HA-tagged full-length NONO construct were generated in pcDNA3.1(+). Constructs were verified by colony PCR, agarose-gel electrophoresis, and Sanger sequencing, and expression was confirmed in HEK-293T cells. Forward and reverse Co-IP defined the binding domains, and binding strength was quantified by densitometry. The SLFN11–NONO interface was modelled by predicting monomer structures with AlphaFold2 (v2.2.0), docking with ZDOCK, and analysing the top-scoring complex in PyMOL (v2.5.4).
2.8 Statistical analysis
Data from at least three independent experiments are presented as mean ± SD. Comparisons between two groups used Student's t-test; multiple groups were compared by one-way ANOVA with appropriate post-hoc tests. P< 0.05 was considered statistically significant.
3Results
3.1 Identification of hAMSCs
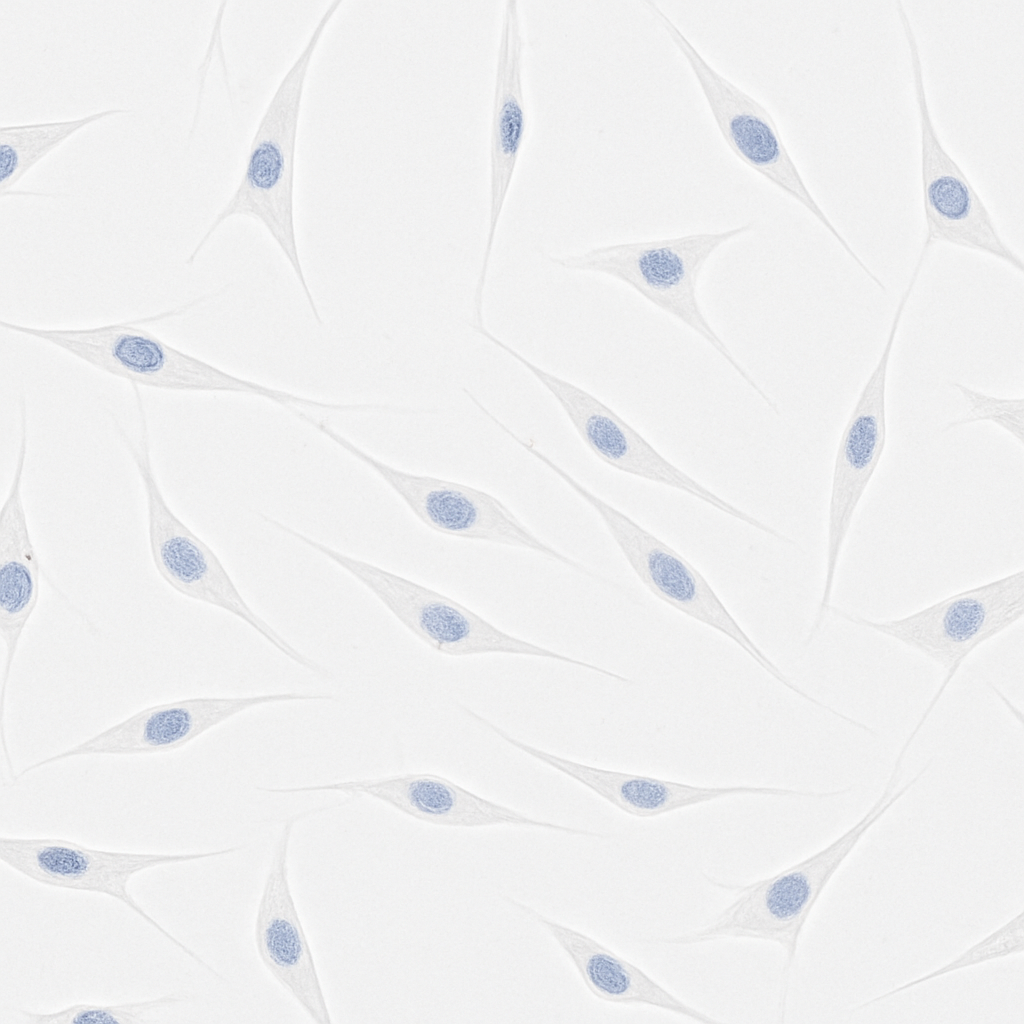
Cells isolated from amniotic membrane by two-step enzymatic digestion displayed a uniform spindle-shaped, fibroblast-like morphology. By immunocytochemistry they were strongly positive for the mesenchymal marker vimentin and negative for the epithelial marker CK19 (Fig. 1A). Flow cytometry confirmed high expression of the MSC surface markers CD105, CD73, CD90, CD44, and CD29 and minimal expression of the haematopoietic/negative cocktail (CD34/CD11b/CD19/CD45/HLA-DR) (Fig. 1B), establishing that the isolated cells were bona fide hAMSCs of high purity suitable for downstream study.
Figure 1. Isolation and phenotypic identification of hAMSCs

B
3.2 Stable overexpression of SLFN11 in hAMSCs
Lentiviral transduction efficiency was optimised by fluorescence intensity, with an MOI of 60 producing >80% infection at the lowest viral dose. Western blot 72 h after transduction confirmed marked upregulation of SLFN11 relative to uninfected (Control) and empty-vector (Mock) cells (Fig. 2A–B). On continuous expansion to passage 18, Control and Mock cells adopted the classic senescent MSC morphology — enlarged, flattened, polygonal cells with slow growth — whereas SLFN11-overexpressing cells retained a slender spindle shape and rapid swirling growth (Fig. 2C), confirming that SLFN11 counteracts expansion-induced morphological ageing.
Figure 2. Lentiviral overexpression of SLFN11 preserves hAMSC morphology
A
B
C Passage 18

SLFN11-OE cells (right) maintain a youthful spindle morphology, while Control and Mock cells flatten and enlarge.
3.3 Establishment of a CPT-induced senescence model
To obtain a defined DNA-damage senescence model, hAMSCs were treated with increasing concentrations of CPT. SA-β-gal positivity rose dose-dependently, increasing from 3.89% in untreated cells to 25.23% at 1 µM (Fig. 3A–B). CCK-8 assays showed that viability was essentially preserved up to 1 µM but fell sharply at ≥5 µM (Fig. 3C). Accordingly, 1 µM CPT was selected to induce senescence without overt cytotoxicity in all subsequent experiments.
Figure 3. Dose-dependent CPT induction of hAMSC senescence
A
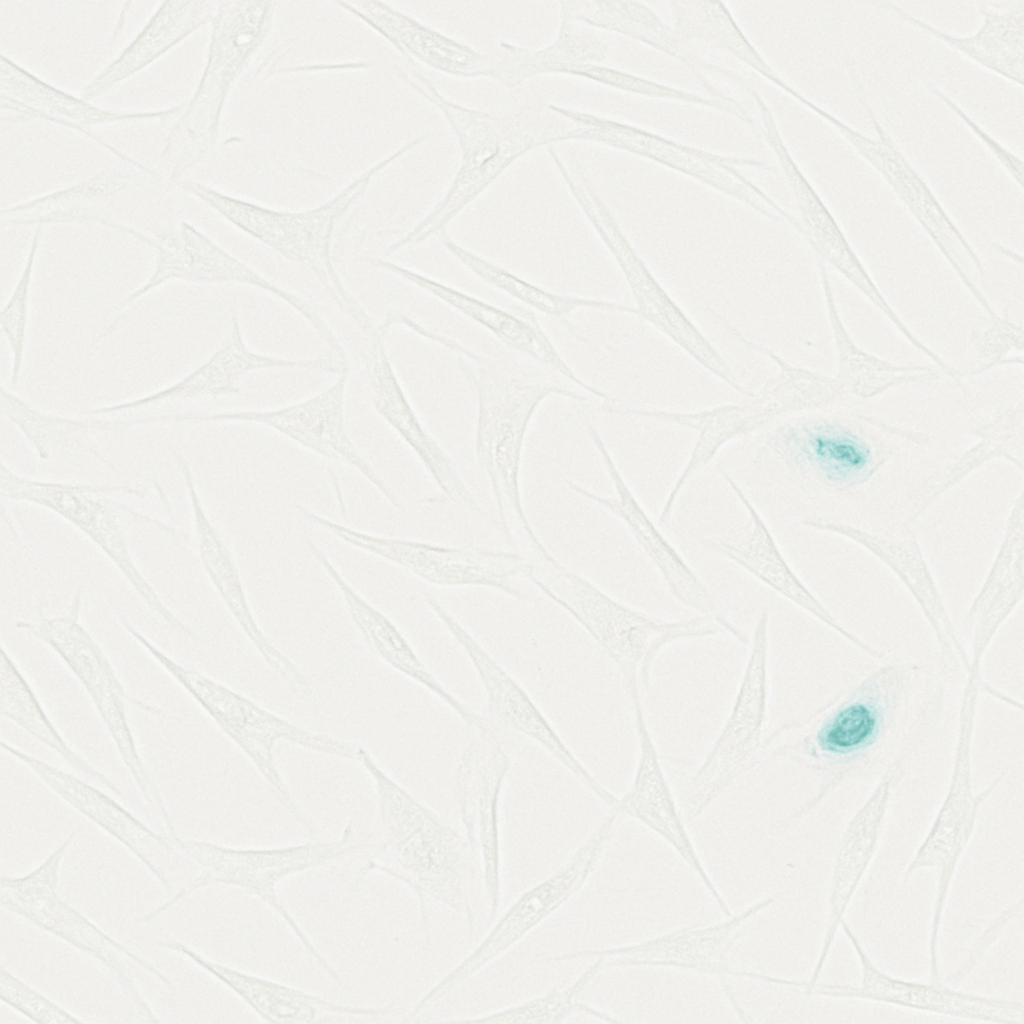
B
C
3.4 Screening of the SLFN11 interactome
To uncover the effector through which SLFN11 acts, IP–MS was performed on CPT-treated SLFN11-overexpressing hAMSCs, detecting 14 proteins uniquely associated with SLFN11 (Fig. 4A). The RNA-binding protein NONO ranked highest (score 39.58), closely followed by DDX1 and G3BP2. Co-IP validation confirmed specific interaction of SLFN11 with NONO, DDX1, G3BP2, and LCN1, but not with several lower-scoring candidates (Fig. 4B). Given its top score and established roles in transcription, DNA-damage repair, and the cell cycle, NONO was prioritised for functional dissection.
Figure 4. SLFN11 interactome and validation of the SLFN11–NONO interaction
A IP–MS scores
B Co-IP validation
3.5 NONO is required for SLFN11-mediated delay of senescence
siRNA efficiently reduced NONO protein levels (Fig. 5A), enabling a rescue design in which NONO was silenced in SLFN11-overexpressing cells. After CPT-induced senescence, SLFN11 overexpression sharply lowered SA-β-gal positivity, but co-silencing NONO restored it to vector-like levels (Fig. 5B). In parallel, SLFN11 overexpression upregulated the stemness factors Nanog, Oct4, and Sox2; this gain was reversed by NONO knockdown (Fig. 5C). The cell-cycle inhibitors p16 and p21 were suppressed by SLFN11 and re-elevated upon NONO silencing (Fig. 5D). Finally, both PCNA immunoblotting and EdU incorporation showed that the proliferative boost conferred by SLFN11 was lost when NONO was depleted (Fig. 5E–F). Thus, SLFN11's anti-senescence activity is functionally dependent on NONO.
Figure 5. NONO knockdown abolishes the anti-senescence effects of SLFN11
A si-NONO
B SA-β-gal
C Stemness factors
D Cell-cycle inhibitors
E PCNA
F EdU proliferation

3.6 Mapping the SLFN11 domain that binds NONO
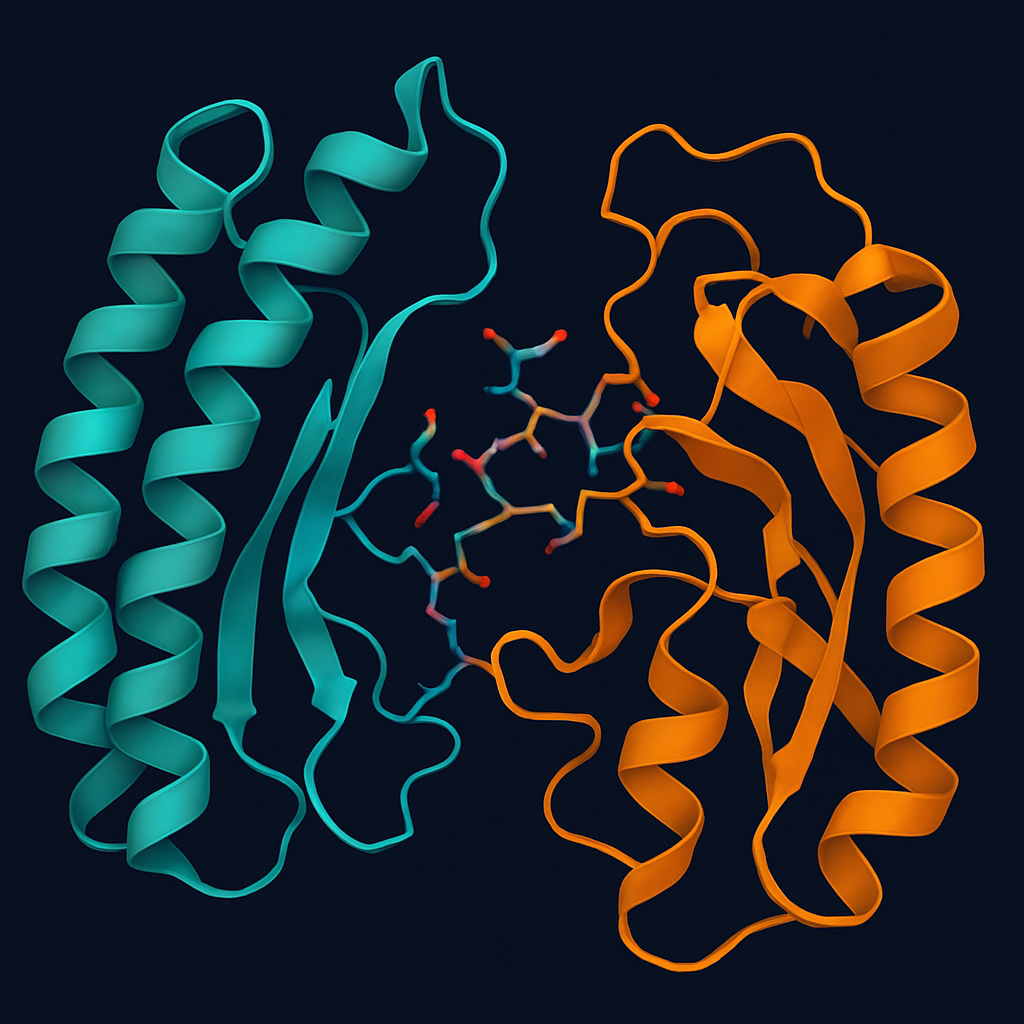
SLFN11 comprises an N-terminal endonuclease domain (1–353 aa), a central linker (354–576 aa), and a C-terminal helicase domain (577–901 aa) (Fig. 6A). Flag-tagged truncations and HA-tagged NONO were cloned, sequence-verified, and confirmed to express at the expected molecular weights in HEK-293T cells (Fig. 6B). Forward Co-IP showed that full-length SLFN11 and all three individual domains co-precipitated NONO, with the C-terminal domain showing the strongest signal (Fig. 7A–B). Reverse Co-IP, using HA-NONO as bait, captured full-length SLFN11 and the N- and C-terminal domains but not the linker (Fig. 8A). Molecular docking (AlphaFold2/ZDOCK) predicted that NONO engages predominantly the long α-helix of the SLFN11 C-terminus through sulfur–π, hydrogen-bond, and electrostatic contacts (Fig. 8B). Together these data localise the functionally dominant SLFN11–NONO interface to the SLFN11 C-terminal domain.
Figure 6. SLFN11 domain architecture and construct validation
A
B Construct expression (HEK-293T)
Figure 7. All SLFN11 domains bind NONO, with the C-terminus strongest
A IP: Flag → IB: HA
B Binding strength
Figure 8. Reverse Co-IP and molecular docking localise NONO binding to the SLFN11 C-terminus
A IP: HA (NONO) → IB: Flag
The linker domain (dimmed) fails to bind NONO in the reverse pulldown, narrowing the functional interface to the N- and, predominantly, C-terminal domains.
B SLFN11–NONO docking model
4Discussion
Building on our earlier observation that SLFN11 protects hAMSCs from DNA-damage accumulation, this study screened 14 candidate SLFN11-interacting proteins by IP–MS and, through Co-IP and rescue experiments (SLFN11 overexpression combined with NONO knockdown), established that NONO is necessary for SLFN11-mediated delay of mesenchymal stem-cell senescence. This not only corroborates the reliability of the SLFN11 anti-senescence phenotype but, for the first time, defines its molecular effector. Notably, silencing NONO reversed every SLFN11-driven senescence readout, suggesting that SLFN11 acts in part by regulating the function or stability of NONO and thereby influencing downstream senescence pathways such as p53/p21 — although the precise signalling cascade awaits further validation.
Ageing is accompanied by a progressive loss of cellular and tissue integrity. Among the established hallmarks of ageing — stem-cell exhaustion, telomere attrition, genomic instability, epigenetic drift, loss of proteostasis, impaired autophagy, deregulated nutrient sensing, mitochondrial dysfunction, altered intercellular communication, chronic inflammation, and dysbiosis — genomic instability is a central driver, and the accumulation of DNA damage promotes ageing by inducing cell death, senescence, and tissue dysfunction. Consistent with our prior finding that SLFN11 overexpression reduces SA-β-gal positivity and apoptosis while enhancing stemness, proliferation, and cell-cycle progression, we here show that each of these protective effects is contingent on NONO. SA-β-gal staining, the canonical senescence assay, and the reciprocal regulation of stemness factors, p16/p21, and PCNA/EdU all moved in concert: SLFN11 ameliorated them and NONO depletion abolished the benefit. SLFN11 therefore exhibits a functional dependence on NONO, the two proteins acting as a co-regulatory complex during hAMSC senescence.
SLFN11, a 901-residue nuclear dimer, was first recognised for restricting viral replication and is now appreciated as a key negative regulator of the DNA-damage response. It contains an N-terminal endonuclease domain (1–353 aa) that degrades type-II tRNAs and down-regulates ATR via a E209/E214/K216 catalytic triad, a central linker of uncertain function (354–576 aa), and a C-terminal helicase domain (577–901 aa) with ssDNA-binding activity that recruits RPA. Our domain-mapping experiments showed that all three truncations expressed cleanly in HEK-293T cells and that each co-precipitated NONO in forward Co-IP, implying multiple contact surfaces. However, reverse Co-IP using NONO as bait captured only the N- and C-terminal domains, and molecular docking predicted that NONO engages chiefly the long α-helix of the C-terminus. We therefore infer that, although several interaction sites exist, the C-terminal helicase domain forms the dominant and functionally relevant interface.
Double-strand breaks are the most cytotoxic DNA lesions and are repaired principally by homologous recombination (HR) and non-homologous end-joining (NHEJ). SLFN11 suppresses HR by binding the RPA1 subunit through its C-terminus and dissociating RPA from ssDNA, and it is recruited to damage sites to maintain chromatin compaction and stall replication forks while blocking ATM/ATR activation. NONO, in turn, is a functional participant in NHEJ: its O-GlcNAcylation (notably at Ser147) stabilises the protein and promotes its recruitment to damage sites, and PARP-1–generated PAR engages the NONO RRM1 domain to drive NHEJ while inhibiting HR. The convergence of SLFN11 and NONO on DNA-damage repair, plausibly via NHEJ, offers a mechanistic rationale for their cooperation, with the SLFN11 C-terminus serving as the principal binding and effector module. The integrated working model is summarised in Fig. 9.
Several limitations merit attention. Co-IP results may be influenced by bridging cofactors in the lysate, so direct interaction between SLFN11 and NONO should be confirmed by in vitro pulldown or cross-linking. Moreover, while the C-terminus most likely mediates NONO binding, we have not yet tested whether the isolated truncations retain anti-senescence activity; transfecting the SLFN11 truncations into hAMSCs to assay senescence markers will be an important next step.
Figure 9. Working model: the SLFN11–NONO axis delays hAMSC senescence
DNA damage
CPT · DSBs
SLFN11
C-terminus
NONO
RNA-binding
Stemness ↑
Proliferation ↑
Arrest ↓
Delayed hAMSC senescence
5Conclusions
In the CPT-induced DNA-damage model, SLFN11 interacts with NONO, DDX1, G3BP2, and LCN1 in hAMSCs. Combining Co-IP, reverse Co-IP, and molecular docking, the SLFN11 C-terminal helicase domain emerges as the most probable site of NONO binding and of anti-senescence function.
NONO is the essential effector through which SLFN11 delays hAMSC senescence, the two proteins forming a functional complex that sustains stemness and proliferation while restraining the p16/p21 cell-cycle–arrest programme. By integrating interactome analysis (IP–MS) with domain mapping, this work provides the first systematic delineation of the structural basis of SLFN11 action and reveals the SLFN11–NONO axis as a candidate regulatory node for delaying tissue ageing and improving the efficacy of stem-cell–based therapies.
Key message
SLFN11 delays the senescence of human amniotic mesenchymal stem cells by engaging the RNA-binding protein NONO — principally through its C-terminal helicase domain — defining the SLFN11–NONO axis as a tractable target for anti-senescence intervention.